Biomolecular Assembly
 Using a range of computational tools including Monte Carlo and Molecular Dynamics simulations
we investigate interactions within and between bio-molecules. Examples are protein-protein
assembly, protein ionization and pKa values, peptide-membrane interactions. We also develop
software for coarse grained protein simulations.
Using a range of computational tools including Monte Carlo and Molecular Dynamics simulations
we investigate interactions within and between bio-molecules. Examples are protein-protein
assembly, protein ionization and pKa values, peptide-membrane interactions. We also develop
software for coarse grained protein simulations.
People:
Mikael Lund,
Marie Skepo, Bo Jonsson
Quantum chemistry software and methodology
 Molecules can have complicated electronic structure, where the
mean-field assumption does not hold, and so-called multiconfiguration
based methods are necessary. This is almost always the case in
photochemistry and spectroscopy, and we are particularly interested
in developing methods to deal with such situations.
Molecules can have complicated electronic structure, where the
mean-field assumption does not hold, and so-called multiconfiguration
based methods are necessary. This is almost always the case in
photochemistry and spectroscopy, and we are particularly interested
in developing methods to deal with such situations.
People: Per Åke Malmqvist, Per-Olof Widmark, Valera Veryazov
Macroscopic Properties of Dipolar Systems
 Our world is built from molecules with properties like charge, size and dipole moment.
The world we experience is characterized by bulk properties like viscosity and dielectric permittivity.
The purpose of this project is to establish a link between the two worlds and investigate how big systems
that are actually needed in order to obtain bulk behavior.
Our world is built from molecules with properties like charge, size and dipole moment.
The world we experience is characterized by bulk properties like viscosity and dielectric permittivity.
The purpose of this project is to establish a link between the two worlds and investigate how big systems
that are actually needed in order to obtain bulk behavior.
People: Gunnar Karlstrom
Salivary Proteins
Salivary proteins are crucial for the oral health. It is important to understand their adsorption
behaviour to prevent caries but also for developing new salivary substitutes and dental products.
We investigate their structural and thermodynamic properties to gain
information about the initial selective adsorption of the proteins in the salivary films.
People: Marie Skepo, Mikael Lund
Theoretical Biochemistry
 We use most types of theoretical methods, ranging from high-level quantum mechanics (QM) methods,
through density functional theory, to molecular mechanics (MM) and statistical mechanics simulation methods.
In particular, we study the structure, function and mechanisms of metalloproteins and the binding of drug candidates
to biomacromolecules.
We also develop software for QM/MM calculations and their combination with experimental techniques
(crystallography, NMR, and EXAFS), as well as for QM/MM free-energy perturbation.
We use most types of theoretical methods, ranging from high-level quantum mechanics (QM) methods,
through density functional theory, to molecular mechanics (MM) and statistical mechanics simulation methods.
In particular, we study the structure, function and mechanisms of metalloproteins and the binding of drug candidates
to biomacromolecules.
We also develop software for QM/MM calculations and their combination with experimental techniques
(crystallography, NMR, and EXAFS), as well as for QM/MM free-energy perturbation.
People: Ulf Ryde
Colloids and Polymers
 We devote considerable efforts to construct theories and simulation techniques, with some emphasis
on classical polymer density functional theory. These are used to study the way in which dissolved
polymers adsorb and mediate interactions in colloidal dispersions.
We devote considerable efforts to construct theories and simulation techniques, with some emphasis
on classical polymer density functional theory. These are used to study the way in which dissolved
polymers adsorb and mediate interactions in colloidal dispersions.
People: Jan Forsman, Bo Jonsson
Theoretical Chemical Physics
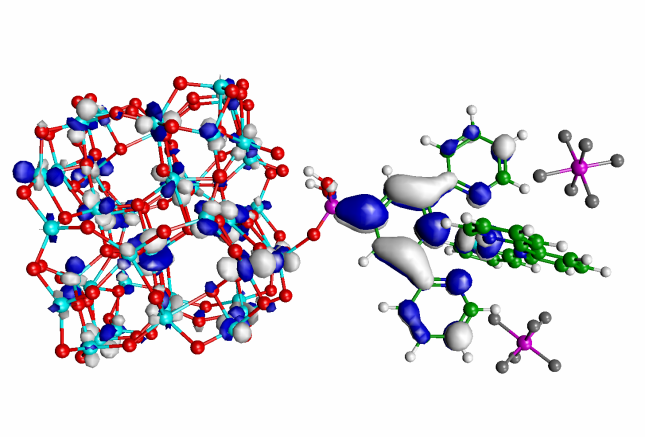 Computational studies of molecules and materials with applications to photochemistry, surface chemistry,
nanostructured materials, and solar energy conversion.
Computational studies of molecules and materials with applications to photochemistry, surface chemistry,
nanostructured materials, and solar energy conversion.
People: Petter Persson
Ionic liquids
We develop classical density functional theories, backed up by simulations, for coarse-grained models
of ionic liquids and salt solutions. These are then our tools to investigate molecular mechanisms
underlying the remarkable physical-chemical behaviour of ionic liquids.
People: Jan Forsman